


Fundamentos de termodinámica
Barbol
Índice General
- 1 Conceptos básicos
- 1.1 Sistema termodinámico
- 1.2 Variables y equilibrio termodinámicos
- 1.3 Procesos e interacción termodinámicos
- 1.4 Máquinas térmicas y frigoríficas
- 2 Temperatura
- 2.1 Equilibrio térmico
- 2.2 Concepto de temperatura: temperatura empírica
- 2.3 Medida de la temperatura. Diferentes termómetros
- 2.4 Termómetro de gas a volumen constante. Termómetro del gas ideal
- 2.5 Estudio particular de algunos termómetros
- 2.6 Escala práctica internacional de temperaturas (1968)
- 3 Sistemas termodinámicos simples. Trabajo termodinámico
- 3.1 Trabajo
- 3.2 Diferentes formas de trabajo cuasiestático
- 3.3 Generalización del trabajo
- 3.4 Coeficientes térmicos
- 4 Calor y primer principio de la termodinámica
- 4.1 Trabajo y energía interna
- 4.2 Formulación matemática del primer principio
- 4.3 Definición termodinámica de calor
- 4.4 Capacidad calorífica
- 5 Segundo principio de la termodinámica
- 5.1 Limitaciones del primer principio
- 5.2 Enunciados clásicos del segundo principio
- 5.3 Ciclo de Carnot. Teoremas de Carnot
- 5.4 Escala termodinámica de temperaturas
- 6 Entropía
- 6.1 Teorema de Clausius
- 6.2 Concepto de entropía
- 6.3 Principio de aumento de entropía. Reversibilidad e irreversibilidad
- 6.4 Ley generalizada de la termodinámica
- 6.5 Trabajo máximo
- 7 Potenciales termodinámicos
- 8 Tercer principio de la termodinámica
- 8.1 Enunciado de Nernst
- 8.2 Consecuencias físicas
- 8.3 Cálculo de entropías absolutas
- 8.4 Inaccesibilidad del cero absoluto
- A. Relaciones entre derivadas parciales
- B. Transformación dual de Legendre
1 Conceptos básicos
La termodinámica surgió como una generalización de los estudios realizados entre la energía mecánica y el calor intercambiados por las máquinas térmicas, y de ahí el nombre de la disciplina. Sin embargo, poco a poco su campo de aplicación se fue ampliando hasta abarcar todos los procesos en los que exista alguna transformación de energía, sea esta del tipo que sea.Como casi toda la física, esta disciplina es fenomenológicia, i.e., se basa en unos principios que no son matemáticamente demostrables, pero que sin embargo son generalizaciones de los estudios experimentales y nunca se ha visto que fallasen.
Para estudiar, pues, la termodinámica, es imprescindible empezar dando algunas definiciones, como por ejemplo cuáles y cómo son los sistemas con los que vamos a tratar y las variables de los que depende.
1.1 Sistema termodinámico
Un sistema termodinámico es cualquier región del espacio sobre la que centramos nuestro interés. Para delimitar esta región la limitamos con una pared (que puede ser real o imaginaria) que la recubre totalmente. La parte del espacio que no forma parte del sistema se denomina entorno o medio. El sistema y el entorno forman el universo.
En termodinámica vamos a estudiar, pues, la evolución de un sistema cuando este interacciona con el entorno que lo rodea, para ello vamos a emplear variables termodinámicas, que no son más que variables que nos dan la información sobre el estado del sistema, el estado dinámico en el que se encuentran las partículas del sistema.
1.2 Variables y equilibrio termodinámicos
Macroscópicamente el estado del sistema se define estudiando un conjunto de propiedades que afectan globalmente al sistema (como el volumen o la carga eléctrica) que denominaremos variables o coordenadas termodinámicas. Hay que hacer notar que las variables termodinámicas son mensurables y que no se necesitan conocer todas las posibles variables que definen un sistema, va a existir un número mínimo de variables que definirán el estado del sistema de forma unívoca, son las variables o coordenadas de estado.
Estas variables se suelen clasificar de dos modos diferentes: por un lado están las variables extrínsecas (que dependen de la naturaleza del sistema y el valor que toman ciertas magnitudes del entorno) e intrínsecas (que sólo dependen de la naturaleza y el estado del sistema); por otro lado tenemos las variables extensivas (las que dependen de la cantidad de materia del sistema) y las intensivas (no dependen de la cantidad de materia del sistema). Es esta última clasificación la más importante y que se empleará a lo largo de los apuntes.
Definimos como magnitud específica a las variables extensivas partidas de una cantidad que nos dé cuenta de la materia del sistema (bien la masa, bien los moles). Es una variable intensiva.
1.3 Procesos e interacción termodinámicos
Decimos que un sistema termodinámico sufre un proceso termodinámico cuando pasa de un estado inicial de equilibrio1 a otro estado final (también de equilibrio). Los diferentes estados por los que pasa el sistema durante el proceso se llaman camino o trayectoria del proceso.
Para que se dé este proceso es necesario que haya interacción entre el sistema y el entorno, y esto sólo puede ocurrir a través de la pared. Básicamente hay tres tipos de interacción: mecánica, másica y térmica.
La interacción mecánica se debe a una variación en las variables extrínsecas (por ejemplo el volumen) y se producirá hasta que las variables intrínsecas asociadas (en el caso del volumen sería la presión) se igualen en el entorno y el medio. En este caso decimos que la pared es adiabática.
La interacción másica se debe al intercambio de materia a través de una pared permeable.
Una interacción térmica es cualquier otro tipo de intercambio de energía. En este caso la pared se denomina diatérmica.
Decimos que el sistema es abierto si pueden existir los tres tipos de interacción, decimos que es cerrado si no se permite el intercambio de materia y decimos que es aislado si no se permite ningún intercambio de energía.
1.4 Máquinas térmicas y frigoríficas
Bajo esta denominación abarcamos a los sistemas que permiten transformar el calor2 en trabajo y viceversa.
Una máquina térmica es el sistema que cede trabajo al medio intercambiando calor a través de sus fronteras de un modo cíclico. Este intercambio de calor lo hace con dos focos caloríficos3, uno caliente y otro frío. En concreto la máquina térmica recibe calor del foco caliente, cede calor al foco frío y suministra trabajo al medio.
La máquina frigorífica es ``lo contrario'' que la máquina térmica. Al recibir trabajo del ambiente toma calor del foco frío y se lo cede al foco caliente. Por supuesto, tanto la máquina térmica como la frigorífica pueden funcionar entre varios focos.
![\includegraphics[width=0.7\textwidth]{maquinas}](img2.png)
Para el estudio de estas máquinas se define el rendimiento, que es la relación entre el beneficio obtenido y el coste. En una máquina térmica la expresión toma la forma
![]() donde
donde ![]() es el trabajo cedido al medio y
es el trabajo cedido al medio y ![]() el calor absorvido de los focos calientes. En una máquina frigorífica la expresión toma la forma
el calor absorvido de los focos calientes. En una máquina frigorífica la expresión toma la forma
![]() donde
donde ![]() es el calor absorvido del foco frío y
es el calor absorvido del foco frío y ![]() el trabajo suministrado por el entorno.
el trabajo suministrado por el entorno.
2 Temperatura
2.1 Equilibrio térmico
2.1.1 Primer postulado de la termodinámica
Un sistema termodinámico aislado termina alcanzando un estado de equilibrio termodinámico que no puede abandonar por si mismo.
2.1.2 Segundo postulado de la termodinámica
El estado de un sistema queda fijado por varias variables extrínsecas y una intrínseca.
Veamos una justificación del segundo postulado de la termodinámica. Si tenemos un sistema termodinámico ![]() en equilibrio y otro sistema termodinámico
en equilibrio y otro sistema termodinámico ![]() aislado del anterior también en equilibrio y los ponemos en contacto a través de una pared diatérmica pueden ocurrir dos cosas, a saber: las variables intrínsecas de los dos sistemas no varían o bien sus variables macroscópicas varían hasta alcanzar otro estado de equilibrio. En este último caso decimos que dos sistemas aislados localmente de su entorno y en contacto diatérmico entre si alcanzan un equilibrio mútuo conocido como equilibrio térmico.
aislado del anterior también en equilibrio y los ponemos en contacto a través de una pared diatérmica pueden ocurrir dos cosas, a saber: las variables intrínsecas de los dos sistemas no varían o bien sus variables macroscópicas varían hasta alcanzar otro estado de equilibrio. En este último caso decimos que dos sistemas aislados localmente de su entorno y en contacto diatérmico entre si alcanzan un equilibrio mútuo conocido como equilibrio térmico.
Hay que hacer notar que como la pared era solamente diatérmica las variables extrínsecas de ![]() y
y ![]() no variaron, sin embargo las variables macroscópicas de estado sí que lo hicieron, esto implica que las variables que definen el estado de un sistema son todas extrínsecas y, por lo menos, una intrínseca. Como es intrínseca y no depende del estado del sistema es una variable intensiva. A esta variable intensiva la denominamos temperatura empírica.
no variaron, sin embargo las variables macroscópicas de estado sí que lo hicieron, esto implica que las variables que definen el estado de un sistema son todas extrínsecas y, por lo menos, una intrínseca. Como es intrínseca y no depende del estado del sistema es una variable intensiva. A esta variable intensiva la denominamos temperatura empírica.
2.1.3 Principio cero de la termodinámica
Si tenemos dos sistemasy
en equilibrio entre si y tenemos también un tercer sistema
en equilibrio con
Esta es la propiedad transitiva o principio cero de la termodinámica.
2.2 Concepto de temperatura: temperatura empírica
Consideremos dos sistemas termodinámicos simples4 ![]() y
y ![]() caracterizados por dos variables, una de ellas extrínseca (
caracterizados por dos variables, una de ellas extrínseca (![]() ) y otra intrínseca (
) y otra intrínseca (![]() ). Por tanto las coordenadas iniciales del sistema
). Por tanto las coordenadas iniciales del sistema ![]() serán
serán
![]() y las del sistema
y las del sistema ![]() serán
serán
![]() . Si los ponemos en contacto mediante una pared diatérmica sus variables intrínsecas irán variando hasta alcanzar el equilibrio térmico. En ese momento podemos designar el equilibrio mediante una relación entre las coordenadas que en forma general se puede escribir como:
. Si los ponemos en contacto mediante una pared diatérmica sus variables intrínsecas irán variando hasta alcanzar el equilibrio térmico. En ese momento podemos designar el equilibrio mediante una relación entre las coordenadas que en forma general se puede escribir como:
Si ahora los aislamos uno del otro, pero los ponemos en contacto diatérmico con un tercer sistema ![]() , por el principio cero obtenemos las relaciones equivalentes:
, por el principio cero obtenemos las relaciones equivalentes:
donde
De las últimas dos ecuaciones podemos despejar
![]() , que tiene que ser físicamente equivalente a la primera expresión para
, que tiene que ser físicamente equivalente a la primera expresión para ![]() , sin embargo ésa expresión no depende de la variable extrínseca del sistema
, sin embargo ésa expresión no depende de la variable extrínseca del sistema ![]() y, por tanto, la expresión se puede reducir a:
y, por tanto, la expresión se puede reducir a:
Aplicando una segunda vez el mismo argumento con los sistemas ![]() y
y ![]() en equilibrio con
en equilibrio con ![]() se obtiene, finalmente, lo siguiente:
se obtiene, finalmente, lo siguiente:
A esa función ![]() se le llama temperatura empírica del sistema. El lugar geométrico de las coordenadas
se le llama temperatura empírica del sistema. El lugar geométrico de las coordenadas ![]() que dan el mismo valor para
que dan el mismo valor para ![]() constituyen una línea isoterma.
constituyen una línea isoterma.
2.3 Medida de la temperatura. Diferentes termómetros
Como la temperatura va a resultar ser una manitud imprescindible en termodinámica tendremos que idear un dispositivo para medirla. Esto se consigue construyendo un sistema patrón al que llamaremos termómetro. Este termómetro lo colocaremos en contacto diatérmico con el sistema de estudio y esperaremos a que se alcance el equilibrio térmico entre los dos sistemas. Cuando ya se consiguió ese equilibrio medimos alguna propiedad macroscópica del sistema (que llamaremos propiedad termométrica) que dependa de la temperatura. Algunos ejemplos de propiedades termométricas son:
- El volumen de un gas a presión constante.
- La presión de un gas a volumen constante.
- La resistencia eléctrica a tensión mecánica constante.
- Fuerza electromotriz a tensión mecánica constante.
- Radiación térmica a altas temperaturas.
- Propiedades magnéticas a bajas temperaturas.
El método empleado en la actualidad5 consiste en relacionar la temperatura empírica con la propiedad termométrica de modo que sean directamente proporcionales, es decir:
Esta relación es arbitraria, y permite establecer la escala6 a partir de un único punto fijo cuya temperatura se decide en convención. El punto fijo elegido es el punto triple del agua, en el que coexisten en el equilibrio agua en forma de sólido, líquido y vapor. El valor asignado a ese punto es ![]() , por tanto:
, por tanto:
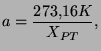
donde

2.4 Termómetro de gas a volumen constante. Termómetro del gas ideal
Se observa experimentalmente que el termómetro de gas es el que presenta diferencias menores con respecto a la temperatua empírica (sobre todo cuanto más rarificado esté el gas), por lo que es el que más se emplea.
El termómetro de gas consiste básicamente en una ampolla que contiene una cantidad fija de gas y se introduce en el sistema a estudiar. Cuando se alcanza el equilibrio térmico se mide la diferencia de alturas entre las dos partes verticales de un tubo en U que contiene mercurio, uno de los lados conectado con la ampolla de gas mediante un tubo capilar. Esta diferencia de alturas nos da la presión a la que se encuentra el gas.
Por supuesto este termómetro presenta algunas inconveniencias que se deben corregir, como gradientes de temperatura en el capilar, dilataciones y contracciones de los contenedores, absorción de gas en las paredes, compresibilidad del mercurio...
Ahora supongamos que estamos calibrando el termómetro. Introducimos una cierta cantidad de gas de modo que el mercurio, en el punto triple, marca una presión ![]() , y en otro sistema genérico marca una presión
, y en otro sistema genérico marca una presión ![]() . Ahora eliminamos algo del gas y obtenemos, para los mismos sistemas, los valores
. Ahora eliminamos algo del gas y obtenemos, para los mismos sistemas, los valores ![]() y
y ![]() . Seguimos reduciendo la cantidad de gas y vamos obteniendo cada vez valores más pequeños para la presión en el punto triple y en el otro sistema. Si ahora representamos
. Seguimos reduciendo la cantidad de gas y vamos obteniendo cada vez valores más pequeños para la presión en el punto triple y en el otro sistema. Si ahora representamos ![]() en función de
en función de ![]() y extrapolamos la curva resultante a
y extrapolamos la curva resultante a ![]() obtenemos que, trabajando con el gas que trabajemos, llegamos al mismo valor
obtenemos que, trabajando con el gas que trabajemos, llegamos al mismo valor
![]() . Por consiguiente la temperatura del gas ideal se define mediante:
. Por consiguiente la temperatura del gas ideal se define mediante:
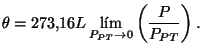
Escala independiente de las propiedades de cualquier gas particular, pero sí de las propiedades generales de los gases. Más adelante (sección 5.4) se demostrará que la escala del gas ideal y la escala termodinámica de temperaturas (que se representa mediante ![]() ) son idénticas.
) son idénticas.
2.5 Estudio particular de algunos termómetros
2.5.1 Termómetro de resistencia eléctrica
La propiedad termométrica es la resistencia eléctrica de un sólido. Esta resistencia se mide manteniendo en el termómetro una corriente constante conocida (ajustada mediante un reostato y comprobada con una resistencia patrón en serie) y midiendo la diferencia de potencial entre sus extremos mediante un voltímetro sensible. A menudo, en un intervalo limitado se emplea la ecuación siguiente para el valor de la resistencia:
donde
2.5.2 Termopar
La propiedad termométrica es la fuerza electromotriz generada por efecto Seebeck. Dos metales soldados con una diferencia de temperaturas entre ellos crean una pequeña fuerza electromotriz. En general la expresión para esta fuerza electromotriz es de la forma:
donde
2.6 Escala práctica internacional de temperaturas (1968)
La Escala Práctica Internacional de Temperaturas (IPTS-68) se compone de una serie de puntos fijos medidos con un termómetro de gas de volumen constante y una colección de procedimientos para la interpolación entre los puntos fijos, de modo que a diferentes intervalos de temperatura podemos usar determinados tipos de termómetro y necesitamos calibrarlos correctamente. Los intervalos de temperatura y los termómetros empleados en ellos son los siguientes:
- Para temperaturas inferiores a
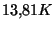 (punto triple del hidrógeno) la escala práctica de temperaturas no está definida, se usan efectos magnéticos para determinar esas temperaturas.
(punto triple del hidrógeno) la escala práctica de temperaturas no está definida, se usan efectos magnéticos para determinar esas temperaturas.
- Desde los
hasta los
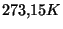 (punto de fusión normal del agua) se emplea un termómetro de resistencia de hilo de platino. En este caso la expresión empleada es
(punto de fusión normal del agua) se emplea un termómetro de resistencia de hilo de platino. En este caso la expresión empleada es
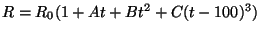 , y por tanto se toman cuatro puntos fijos escogidos de la tabla de puntos fijos de la IPTS-68 para su calibrado.
, y por tanto se toman cuatro puntos fijos escogidos de la tabla de puntos fijos de la IPTS-68 para su calibrado.
- Desde
hasta
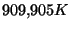 (punto de fusión normal del antimonio) se emplea también el termómetro de resistencia de platino, pero esta vez con su expresión cuadrática. Para el calibrado se emplean el punto triple del agua, el punto de ebullición normal del agua y el punto de fusión normal del cinc.
(punto de fusión normal del antimonio) se emplea también el termómetro de resistencia de platino, pero esta vez con su expresión cuadrática. Para el calibrado se emplean el punto triple del agua, el punto de ebullición normal del agua y el punto de fusión normal del cinc.
- Desde
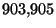 hasta
hasta 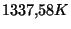 (punto de fusión normal del oro) se emplea un termopar con un hilo de platino de pureza especificada y el otro de Pt(90%)/Rd(10%), con una soldadura mantenida a
(punto de fusión normal del oro) se emplea un termopar con un hilo de platino de pureza especificada y el otro de Pt(90%)/Rd(10%), con una soldadura mantenida a
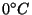 . Para calibrarlo se emplea el punto de fusión normal del antimonio, el punto de fusión normal de la plata y el punto de fusión normal del oro.
. Para calibrarlo se emplea el punto de fusión normal del antimonio, el punto de fusión normal de la plata y el punto de fusión normal del oro.
- Para temperaturas superiores a
la escala tampoco está definica, se usan pirómetros ópticos de radiación monocromática para estimar la temperatura.
3 Sistemas termodinámicos simples. Trabajo termodinámico
3.1 Trabajo
Sobre nuestros sistemas termodinámicos puede actuar una fuerza determinada7 que provoque un desplazamiento. En este caso se realizará un trabajo que vendrá dado por:
El símbolo
![]() implica que esta no es una diferencial exacta, sinó que depende del camino realizado para ir del estado inicial al final. Por este motivo la integral no nos da el valor
implica que esta no es una diferencial exacta, sinó que depende del camino realizado para ir del estado inicial al final. Por este motivo la integral no nos da el valor ![]() sinó el trabajo neto intercambiado.
sinó el trabajo neto intercambiado.
Nos interesa, pues, expresar ese trabajo en función de las variables de estado que nos determinan el estado del sistema. Llamaremos trabajo externo al trabajo realizado por la resultante de todas las fuerzas que el medio ejerce sobre el sistema y trabajo interno al trabajo realizado por las fuerzas ejercidas entre diversas partes del sistema. Es el trabajo externo el que nos interesa estudiar, pues nos dará información acerca del intercambio de energía a través de la frontera del sistema.
Por tanto tenemos que el trabajo es un intercambio de energía a través de la pared, es decir, no es una energía almacenada por el sistema y por tanto no es una característica del estado del mismo, i.e., no es variable o función de estado.
En termodinámica se adopta el mismo criterio de signos que en mecánica (consensuado por la IUPAC en 1970). Si el sistema gana energía se considera que el trabajo es positivo, y si el sistema pierde energía (es decir, la gana el medio) se considera que el trabajo es negativo.
3.2 Diferentes formas de trabajo cuasiestático
Damos a continuación la expresión correspondiente al trabajo de varios sistemas simples y de uso importante en la termodinámica.
3.2.1 Sistema hidrostático o expansivo
En este caso el sistema interacciona con el medio a través de una pared que sólo permite una variación de volumen. El medio ejercerá una presión uniforme sobre toda la superficie de modo que ésta cambia de forma y tamaño.
Sea ![]() la presión ejercida por el medio y
la presión ejercida por el medio y
![]() la variación infinitesimal de volumen del sistema, en ese caso:
la variación infinitesimal de volumen del sistema, en ese caso:
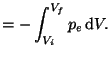 |
Si el proceso tiene lugar de forma cuasiestática las fuerzas interna y externa son casi iguales y podemos considerar que ![]() , siendo
, siendo ![]() la presión del sistema, por tanto:
la presión del sistema, por tanto:
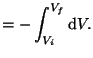 |
El signo aparece para ser consecuente con el criterio de signos empleado. En este sistema la presión es la variable intensiva y el volumen la variable extensiva, de modo que podemos expresar el trabajo simplemente en las coordenadas ![]() .
.
3.2.2 Trabajo al variar la longitud de un alambre
Si tenemos un alambre que por causa de una tracción sufre una elongación (y suponiendo que la temperatura permanezca constante) entonces obtenemos la expresión:
donde
En este sistema la fuerza es la variable intensiva y la elongación la variable extensiva, de modo que podemos expresar el trabajo simplemente en las coordenadas ![]() . Generalmente se emplea el módulo de Young isotérmico (
. Generalmente se emplea el módulo de Young isotérmico (
![]() ) siendo
) siendo ![]() la sección del hilo a la hora de hacer cálculos con este tipo de sistema.
la sección del hilo a la hora de hacer cálculos con este tipo de sistema.
3.2.3 Trabajo al variar el área de una lámina superficial
Supongamos una lámina superficial de conteniendo líquido (como por ejemplo una película de jabón) extendida en un armazón con un lado móvil, en ese caso el trabajo infinitesimal es:
donde
En este sistema la tensión superficial es la variable intensiva y el área la variable extensiva, de modo que podemos expresar el trabajo simplemente en las coordenadas
![]() .
.
3.2.4 Trabajo eléctrico de una pila
Una pila produce una corriente eléctrica como consecuencia de una reacción química controlada. Se crea una diferencia de potencial
![]() que provoca un desplazamiento de las cargas. Por tanto el trabajo es:
que provoca un desplazamiento de las cargas. Por tanto el trabajo es:
En este sistema la fuerza electromotriz es la variable intensiva y la carga la variable extensiva, de modo que podemos expresar el trabajo simplemente en las coordenadas
![]() .
.
3.2.5 Trabajo al polarizar un dieléctrico
Cuando aplicamos un campo eléctrico sobre un dieléctrico las cargas positivas y las negativas de las moléculas se desplazan creando dipolos. Estos dipolos crean un pequeño campo eléctrico denominado campo eléctrico de polarización. Si consideramos que el medio es isótropo y homogéneo la expresión del trabajo es:
donde
En este sistema el campo eléctrico es la variable intensiva y la polarización la variable extensiva, de modo que podemos expresar el trabajo simplemente en las coordenadas
![]() .
.
3.2.6 Trabajo al magnetizar una sustancia paramagnética
Al someter una sustancia paramagnética a la acción de un campo magnético ![]() esa sustancia orienta sus momentos dipolares magnéticos variando entonces su magnetización. Suponiendo que la sustancia sea isótropa y homogénea la expresión del trabajo es:
esa sustancia orienta sus momentos dipolares magnéticos variando entonces su magnetización. Suponiendo que la sustancia sea isótropa y homogénea la expresión del trabajo es:
donde
En este sistema el campo magnético es la variable intensiva y la magnetización la variable extensiva, de modo que podemos expresar el trabajo simplemente en las coordenadas
![]() .
.
3.3 Generalización del trabajo
Todas las ecuaciones son de la forma
![]() , con
, con ![]() la variable intensiva y
la variable intensiva y
![]() la variación que experimenta la variable de deformación. A la variable intensiva también se le llama fuerza generalizada.
la variación que experimenta la variable de deformación. A la variable intensiva también se le llama fuerza generalizada.
Si el trabajo tiene lugar a través de varias variables de deformación el trabajo total sería:
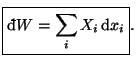
3.4 Coeficientes térmicos
El estado de un sistema está determinado por las variables de estado. Cualquier magnitud que dependa del estado del sistema variará cuando se produzca una variación de cualquiera de estas variables.
Midiendo y tabulando los coeficientes térmicos para los diferentes sistemas podemos facilitar el trabajo de investigación, y por eso es útil definirlos.
Supongamos que un sistema simple tiene un estado dado por una variable extensiva y la temperatura (coordenadas ![]() ), entonces definimos la magnitud de estado
), entonces definimos la magnitud de estado
![]() . Los diferentes coeficientes térmicos los definimos a continuación.
. Los diferentes coeficientes térmicos los definimos a continuación.
3.4.1 Coeficiente de dilatación térmica
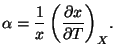
Nos indica cómo varía la variable de deformación al variar la temperatura manteniendo constante la fuerza generalizada y por unidad de variable de deformación. Hay que hacer notar que
![]() .
.
3.4.2 Coeficiente pezométrico
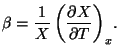
Nos indica cómo varía la fuerza generalizada al variar la temperatura manteniendo constante la variable de deformación y por unidad de fuerza generalizada. Hay que hacer notar que
![]() .
.
3.4.3 Coeficiente de compresibilidad isotérmico
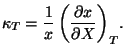
En este caso
![]() .
.
3.4.4 Relación entre los coeficientes
A partir de esas definiciones y con las relaciones que se muestran en el apéndice A buscamos la relación entre ![]() ,
, ![]() y
y
![]() . Esto es útil ya que normalmente
. Esto es útil ya que normalmente ![]() es complicado de medir experimentalmente y así podemos determinarlo a partir de los otros dos coeficientes:
es complicado de medir experimentalmente y así podemos determinarlo a partir de los otros dos coeficientes:
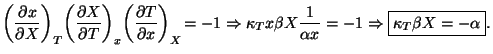
4 Calor y primer principio de la termodinámica
Los principios son generalizaciones fruto de muchos trabajos empíricos que generalmente introducen nuevas magnitudes. El primer principio de la termodinámica parte de la comprobación experimental de que un sistema puede sufrir un cambio desde un estado inicial a un estado final a través de un proceso adiabático.
4.1 Trabajo y energía interna
Supongamos que tenemos un sistema consistente en un fluído que llena un recipiente cerrado con una pared adiabática. Junto con el fluído podemos tener una rueda de paletas que se mueva mediante un mecanismo externo. Por tanto, existe un intercambio adiabático de trabajo mecánico y se comprueba que la temperatura varía.
De igual modo supongamos que tenemos el mismo recipiente con el mismo líquido, pero esta vez en lugar de una rueda de paletas el sistema está compuesto también por una resistencia. Si hacemos pasar una corriente eléctrica a través de la resistencia observaremos que la temperatura varía y que el proceso sigue siendo adiabático (el sistema es el fluído con la resistencia).
Experimentalmente se comprobó que el trabajo realizado para ir desde el mismo estado inicial al mismo estado final es el mismo si se realiza adiabáticamente, a partir de lo cual se define el primer principio de la termodinámica:
Siempre es posible que un sistema sufra un proceso adiabático entre dos estados de equilibrio dados y, sea cual sea el tipo de proceso, el trabajo intercambiado adiabáticamente entre los mismos estados toma el mismo valor.
Este postulado es muy importante porque nos introduce la noción de energía interna: siempre que se encuentra una magnitud que depende sólo de los estados inicial y final, y no de la trayectoria que los une, es posible encontrar una función cuyo valor final menos su valor inicial es igual al trabajo realizado8. En el caso de la termodinámica es posible encontrar una función de las coordenadas del sistema cuyo valor en el estado final menos su valor en el estado inicial es igual al trabajo adiabático intercambiado, es la función energía interna ![]() . Por tanto tenemos que
. Por tanto tenemos que
Esa energía interna no es más que la suma de todas las energías que poseen las partículas que componen el sistema. Como se puede observar es una función de estado extensiva e inherente al sistema: no podemos hablar del trabajo que posee un sistema y, como se verá más adelante, tampoco del calor que posee, sin embargo sí que podemos hablar con propiedad de la energía interna de un sistema.
La diferencia ![]() se interpreta físicamente como la variación de energía del sistema, por tanto la igualdad de la variación de la energía y del trabajo adiabático expresa el principio de conservación de la energía, además de la existencia de la función energía interna como tal.
se interpreta físicamente como la variación de energía del sistema, por tanto la igualdad de la variación de la energía y del trabajo adiabático expresa el principio de conservación de la energía, además de la existencia de la función energía interna como tal.
Además la energía interna es una función de tantas coordenadas termodinámicas como son necesarias para especificar el estado de un sistema. Por ejemplo, en un sistema hidrostático podemos expresar la función energía interna como función de dos cualesquiera de las coordenadas ![]() ,
, ![]() y
y ![]() , estando la otra coordenada fijada por la ecuación de estado.
, estando la otra coordenada fijada por la ecuación de estado.
4.2 Formulación matemática del primer principio
Se acaba de definir la diferencia de energía interna entre dos estados de un sistema como el trabajo adiabático empleado para pasar de un sistema a otro; por otra parte ya adelantamos que el calor es la energía que intercambia el sistema con el medio debido a una diferencia de temperaturas. Supongamos ahora que el sistema pasa de un estado a otro por medio de un proceso no adiabático. Si no es adiabático el sistema intercambiará calor y trabajo con el medio. Como la energía interna es función de estado la variación de energía interna será igual yendo por una trayectoria adiabática que por una que no es adiabática, por tanto tenemos:
que es la expresión mas general para el primer principio de la termodinámica.
Vemos que el calor intercambiado por el sistema es la diferencia entre el trabajo intercambiado adiabáticamente y el trabajo intercambiado en el mismo proceso que el calor, por tanto, como el trabajo no es función de estado y tanto la energía interna como el trabajo son magnitudes extensivas, el calor es una magnitud extensiva que depende del proceso (i.e., no es función de estado).
Otra forma muy usual para el primer principio de la termodinámica es su forma diferencial:
4.3 Definición termodinámica de calor
El calor es energía en tránsito desde un sistema al medio debido a una diferencia de temperaturas. Durante el proceso no se conoce el calor, sólo se conoce la velocidad de flujo de calor, así que debemos esperar a que haya transcurrido un tiempo para poder decir cuánto calor ha sido transferido de un sistema al otro.
Por tanto, si tenemos dos sistemas en contacto y, en su conjunto, aislados adiabáticamente, el calor perdido por un sistema es igual al calor ganado por el otro.
Además el primer principio permite transformar cualquier tipo de energía en energía térmica y viceversa, sin embargo impide la existencia del móvil perpétuo de primera especie: una máquina que produzca trabajo de la nada o que produzca más trabajo que el calor que extrae.
4.4 Capacidad calorífica
Las propiedades térmicas de un sistema termodinámico se miden mediante los coeficientes térmicos, ya estudiados en la sección 3.4.
Las propiedades energéticas nos indican cómo varía la energía interna del sistema al variar alguna de las variables de estado, o cómo varía la energía intercambiada en forma de calor al variar las variables de estado.
4.4.1 Capacidad calorífica
Supongamos un sistema intercambiando calor con el medio. Por tanto la temperatura del sistema sufre una variación ![]() . Se define la capacidad calorífica del sistema como:
. Se define la capacidad calorífica del sistema como:
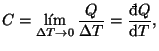
que no es una derivada, sinó un cociente de infinitesimales. Esta magnitud es extensiva y por tanto podemos definir la capacidad calorífica específica (por unidad de masa o mol) dividiéndola por
La capacidad calorífica a variable de deformación constante se expresa
![]() y la capacidad calorífica a fuerza generalizada constante se expresa
y la capacidad calorífica a fuerza generalizada constante se expresa
![]() .
.
4.4.2 Calores latentes
Los calores latentes son una medida del calor intercambiado cuando varía la variable de deformación o la fuerza generalizada en un proceso isotermo. Las expresiones son las siguientes: para el calor latente variando la variable de deformación
![]() , para el calor latente variando la fuerza generalizada
, para el calor latente variando la fuerza generalizada
![]() .
.
4.4.3 Calores latentes de transición
Hasta ahora hablamos de sistemas en una sola fase, pero puede suceder que al intercambiar energía térmica el sistema cambie de fase (por ejemplo de sólido a líquido) detectable por la variación de sus variables físicas. En un sistema hidrostático las transiciones de fase tienen lugar a presión y temperatura constantes, mientras dura la transición el agua pasa a hielo sin que varíe ni la temperatura ni la presión del sistema. El calor latente de transición es el calor intercambiado por unidad de masa mientras dura la transición:
![]() .
.
5 Segundo principio de la termodinámica
5.1 Limitaciones del primer principio
El primer principio de la termodinámica establece que la energía se conserva cuando un sistema pasa de un estado inicial a un estado final, pero nada nos dice de cómo podemos saber cuál de los dos estados es el inicial y cual el final, i.e., no nos da información acerca de la evolución del sistema. Por tanto el primer principio es insuficiente para indicarnos el estado de un sistema.
Por ejemplo, si juntamos dos gases uno a una temperatura mayor que otra no hay nada en el primer principio que impida que el gas caliente se caliente más y el gas frío se enfríe, sin embargo sabemos por la experiencia que este nunca es el caso. A partir de experiencias como ésta deducimos un nuevo principio que implica una nueva variable de estado.
5.2 Enunciados clásicos del segundo principio
El segundo principio de la termodinámica es una generalización de un comportamiento experimental que se hizo a partir del estudio de los conversores de calor, y se suele dar en dos enunciados que resultan ser equivalentes, como se demostrará en la sección 5.2.3.
5.2.1 Primer enunciado (Kelvin-Planck)
No existe una máquina térmica que extraiga calor de un único foco y suministre trabajo al medio sin ceder calor a otros focos.
![\includegraphics[width=0.35\textwidth]{kelvinplanck}](img124.png)
Como consecuencia de este enunciado observamos que
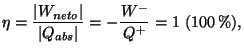
es decir, no existe la máquina térmica de rendimiento unidad.
5.2.2 Segundo enunciado (Clausius)
No existe una máquina frigorífica que extraiga calor del foco frío para cederlo exclusivamente al foco caliente.
![\includegraphics[width=0.35\textwidth]{clausius}](img126.png)
Como consecuencia de este enunciado observamos que
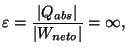
es decir, no existe la máquina frigorífica de eficiencia infinita.
5.2.3 Equivalencia entre los enunciados de Kelvin-Planck y Clausius
Supongamos que tenemos una máquina que viola el enunciado de Kelvin-Planck (que notaremos sin primar) y una máquina frigorífica normal (que notaremos primada) trabajando entre los mismos focos a ![]() el foco caliente y
el foco caliente y ![]() el foco frío. En ese caso tendremos las relaciones:
el foco frío. En ese caso tendremos las relaciones:
Si ahora acoplamos ambas máquinas de modo que el trabajo que realiza la máquina que viola el enunciado de Kelvin-Planck es el trabajo que recibe la máquina frigorífica tenemos que
![]() , entonces sumando las dos ecuaciones anteriores obtenemos:
, entonces sumando las dos ecuaciones anteriores obtenemos:
que no es más que una máquina que viola el enunciado de Clausius. Se puede demostrar análogamente que si conectamos una máquina que viola el enunciado de Clausius y una máquina térmica normal el resultado es una máquina que viola el enunciado de Kelvin-Planck.
Además, vemos que el segundo principio de la termodinámica no permite la existencia de máquinas perpétuas de segunda especie, es decir, aquellas que pueden obtener energía continuamente de un foco frío para realizar trabajo en un foco caliente sin coste.
5.3 Ciclo de Carnot. Teoremas de Carnot
Para poder llegar a una formulación matemática del segundo principio de la termodinámica empleamos lo que se conoce como un ciclo de Carnot: una máquina que funciona cíclicamente entre dos focos mediante procesos reversibles, por tanto puede ser motor o frigorífico. Un ciclo de Carnot está constituido por dos transformaciones isotermas reversibles y dos transformaciones adiabáticas reversibles. Durante las transformaciones isotermas el sistema absorve y cede calor a temperatura constante y en las transformaciones adiabáticas el sistema intercambia trabajo.
5.3.1 Primer teorema de Carnot
El rendimiento de una máquina de Carnot que funcione entre dos focos térmicos es superior al de cualquier máquina real funcionando entre los mismos puntos.
La forma de demostrar este teorema es poniendo dos máquinas a trabajar entre los mismos focos, siendo una real y otra una máquina de Carnot. Si las acoplamos de alguna manera y aplicamos el primer principio vemos que la única forma de que no se viole ninguno de los enunciados del segundo principio implica que el rendimiento de la máquina de Carnot es superior al de la máquina real.
5.3.2 Segundo teorema de Carnot
Cualquier máquina reversible que funcione entre los mismos focos tiene el mismo rendimiento sea cual sea la sustancia con la que trabaja.
La demostración de este teorema es que si tuviesen un rendimiento diferente, al invertir una de las máquinas se podría acoplar con la otra dando lugar a la violación de alguno de los enunciados del segundo principio.
5.4 Escala termodinámica de temperaturas
Se puede demostrar9 que el cociente
![]() es función de la temperatura de los focos, y más aún:
es función de la temperatura de los focos, y más aún:
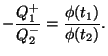
Podemos suponer que
![]() , es decir, una constante por la temperatura termodinámica del foco que, por convenio, se considera positiva. Por tanto tenemos que
, es decir, una constante por la temperatura termodinámica del foco que, por convenio, se considera positiva. Por tanto tenemos que
![]() . Además se observa que la temperatura del foco caliente es mayor que la temperatura del foco frío.
. Además se observa que la temperatura del foco caliente es mayor que la temperatura del foco frío.
Para que coincidan numéricamente las temperaturas termodinámica y del gas ideal en el punto triple se le asigna a la temperatura en ese punto el valor
![]() , aunque el concepto es diferente.
, aunque el concepto es diferente.
Definiendo la temperatura en el punto triple, se puede comprobar fácilmente que la temperatura termodinámica y la del gas ideal coinciden en todo el rango de valores.
6 Entropía
Al igual que el primer principio de la termodinámica nos llevó a postular la existencia de una nueva magnitud denominada energía interna del sistema que nos daba una relación entre el trabajo realizado en el sistema y el calor intercambiado con el medio, el segundo principio nos conduce a una nueva magnitud que denominaremos entropía y que nos indicará qué procesos se pueden dar en la naturaleza de modo espontáneo y cuáles no. Para definir esta magnitud partimos del teorema de Clausius.
6.1 Teorema de Clausius
Se ha dicho que en el caso de las máquinas reversibles existe la relación
![]() , lo cual implica que, de forma genérica,
, lo cual implica que, de forma genérica,
![]() .
.
Ahora generalizaremos esta expresión para el caso de un sistema que trabaje intercambiando calor con un número de focos ![]() a temperatura
a temperatura ![]() . El sistema intercambia calor (
. El sistema intercambia calor (![]() ) con esos focos y, a su vez, esos focos intercambian calor (
) con esos focos y, a su vez, esos focos intercambian calor (![]() ) mediante máquinas de Carnot con un foco común a temperatura
) mediante máquinas de Carnot con un foco común a temperatura ![]() , de modo que el calor que el sistema intercambia con cada uno de los focos es igual pero de signo contrario al que la máquina de Carnot correspondiente intercambia con él (
, de modo que el calor que el sistema intercambia con cada uno de los focos es igual pero de signo contrario al que la máquina de Carnot correspondiente intercambia con él (
![]() ). Además el sistema puede, en general, producir o recibir trabajo (
). Además el sistema puede, en general, producir o recibir trabajo (![]() ) y las máquinas de Carnot un trabajo
) y las máquinas de Carnot un trabajo ![]() .
.
Partiendo de todo esto vamos a llegar a la expresión del calor total intercambiado con el foco común:
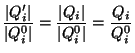 |
||
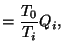 |
||
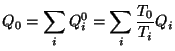 |
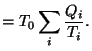 |
Por tanto, vemos que el sistema equivale a una sola máquina que intercambia calor ![]() con un solo foco a temperatura
con un solo foco a temperatura ![]() , con un trabajo resultante igual a
, con un trabajo resultante igual a
![]() . Tenemos, pues, dos posibilidades, la primera es que la máquina tome calor de un foco caliente
. Tenemos, pues, dos posibilidades, la primera es que la máquina tome calor de un foco caliente ![]() y ceda un trabajo al medio, pero esta máquina violaría el enunciado de Kelvin-Planck. La otra posibilidad es una máquina que reciba trabajo del medio (
y ceda un trabajo al medio, pero esta máquina violaría el enunciado de Kelvin-Planck. La otra posibilidad es una máquina que reciba trabajo del medio (![]() ) y ceda calor a un foco frío (
) y ceda calor a un foco frío (
![]() ).
).
A partir de la igualdad anterior y este resultado, y dado que las temperaturas están definidas positivas tenemos la relación
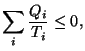
en donde la igualdad se cumple para los procesos reversibles (ya que al invertir el funcionamiento de la máquina cambiarían los signos de los calores y por tanto la suma es nula o positiva) y la desigualdad en los procesos reales.
Si extendemos la desigualdad a un número infinito de focos el sumatorio se convierte en:
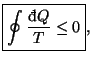
conocida como desigualdad o teorema de Clausius. La integral es cerrada porque es para cada ciclo que sufra el sistema. La igualdad se cumple sólo en los procesos reversibles.
6.2 Concepto de entropía
Supongamos un sistema que evoluciona cíclicamente entre dos estados de equilibrio ![]() y
y ![]() por medio de dos procesos reversibles
por medio de dos procesos reversibles ![]() y
y ![]() . Según el teorema de Clausius, y como los procesos son reversibles:
. Según el teorema de Clausius, y como los procesos son reversibles:
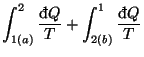 |
||
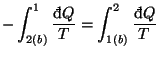 |
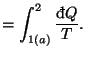 |
Vemos que las dos integrales coinciden sigamos el camino que sigamos, sólo dependen del estado inicial y del estado final, o lo que es lo mismo10, cada una de esas integrales representa la variación de una magnitud que depende sólo del estado del sistema y que denominamos entropía del sistema:
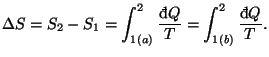
La entropía es una función de las variables de estado del sistema tal que su variación coincide con el calor intercambiado de modo reversible partido por la temperatura:
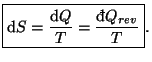
La entropía es una magnitud extensiva. Si el proceso es reversible y adiabático se observa que también es isoentrópico.
Ahora supongamos que vamos entre los estados ![]() y
y ![]() a través de un proceso real, la diferencia de entropías es
a través de un proceso real, la diferencia de entropías es
![]() , la misma que la calculada en un proceso reversible, pero en este caso el calor intercambiado no es el mismo y por tanto
, la misma que la calculada en un proceso reversible, pero en este caso el calor intercambiado no es el mismo y por tanto
![]() .
.
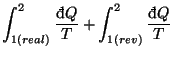 |
||
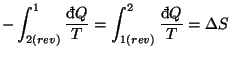 |
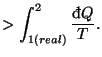 |
Por tanto la variación de entropía siempre es mayor que el trabajo intercambiado partido por la temperatura en procesos reales, y también se observa que el calor intercambiado de forma reversible es siempre mayor que el calor intercambiado en procesos reales.
6.2.1 Coeficiente de compresibilidad adiabático
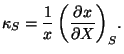
En este caso
![]() .
.
6.3 Principio de aumento de entropía. Reversibilidad e irreversibilidad
En un proceso adiabático
![]() y además
y además
![]() , la cual es la desigualdad de Clausius para procesos adiabáticos. Si además el proceso es reversible se cumple la igualdad.
, la cual es la desigualdad de Clausius para procesos adiabáticos. Si además el proceso es reversible se cumple la igualdad.
Supongamos ahora un sistema aislado (
![]() ). En ese caso también se cumple la desigualdad de Clausius:
). En ese caso también se cumple la desigualdad de Clausius:
que se conoce como principio de aumento de la entropía o expresión general del segundo principio, nos indica que un sistema aislado sólo puede evolucionar espontáneamente hacia estados de entropía creciente. Por tanto, un sistema en equilibrio estable está en un estado de máxima entropía e implica que existen estados del sistema que son inaccesibles por vía adiabática (todos aquellos que impliquen una disminución de entropía). Por tanto podemos corregir el primer enunciado con el segundo principio diciendo:
Ningún estado de un sistema que implique una disminución de entropía cuando se pasa del estado inicial a un estado final se pueden alcanzar por vía adiabática.
El Universo es, globalmente, un sistema aislado, por tanto tenemos que su variación de entropía siempre ha de aumentar o ser nula, pero
![]() . Es decir, un sistema puede sufrir un proceso cualquiera y ser su variación de entropía mayor, menor o igual que cero, y lo mismo se cumple para el medio, pero sin embargo su suma debe ser positiva o nula.
. Es decir, un sistema puede sufrir un proceso cualquiera y ser su variación de entropía mayor, menor o igual que cero, y lo mismo se cumple para el medio, pero sin embargo su suma debe ser positiva o nula.
En un foco térmico se tiene que
![]() , en donde se da la igualdad porque sólo intercambia calor con el sistema (que en este caso sería el medio) y su temperatura permanece constante, por tanto está siempre en equilibrio y el proceso es reversible.
, en donde se da la igualdad porque sólo intercambia calor con el sistema (que en este caso sería el medio) y su temperatura permanece constante, por tanto está siempre en equilibrio y el proceso es reversible.
6.4 Ley generalizada de la termodinámica
Podemos combinar las expresiones matemáticas del primer y el segundo principio de la termodinámica del siguiente modo:
| (1) |
6.5 Trabajo máximo
Supongamos un sistema que sufra un proceso infinitesimal cualquiera entre dos estados infinitamente próximos, entonces:
Si además el proceso es reversible tenemos que:
y si es irreversible
Igualando las dos ecuaciones obtenemos:
Considerando las dos últimas ecuaciones llegamos a:
Por tanto, el trabajo que realiza un sistema en un proceso irreversible es siempre menor que el trabajo que realiza ese mismo sistema en un proceso reversible, i.e., el máximo trabajo realizable por un sistema se da cuando la transformación sufrida es reversible.
7 Potenciales termodinámicos
Durante todos estos apuntes se tuvieron en cuenta las ecuaciones fundamentales de un sistema en representación energía y, en el último capítulo, en representación entropía. En estas dos formas las variables independientes son extensivas
![]() ó
ó
![]() , en tanto que las magnitudes intensivas se obtienen derivando la expresión correspondiente.
, en tanto que las magnitudes intensivas se obtienen derivando la expresión correspondiente.
Sin embargo, en la práctica las variables extensivas suelen ser complicadas de determinar, en tanto que las variables intensivas son fácilmente mensurables. Siendo esto así conviene transformar las ecuaciones fundamentales de un modo que no perdamos información y obtener así unas nuevas funciones que sean función de magnitudes intensivas, éstos serán los potenciales termodinámicos.
7.1 Los diferentes potenciales termodinámicos
Presentamos aquí una lista de los potenciales termodinámicos más usuales y sus expresiones matemáticas útiles, para una justificación matemática general de los mismos véase el apéndice B.
Aquí nos restringiremos a los sistemas con solo una variable de deformación.
7.1.1 Potencial de Helmholtz (energía libre)
Es la transformada de Legendre considerando como variable activa la entropía, por tanto tenemos que:
La ecuación de Euler correspondiente a este potencial es:
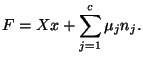
La ecuación de Gibbs-Duhem es:
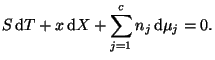
Y las correspondientes relaciones de Maxwell son:
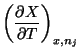 |
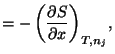 |
|
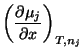 |
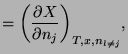 |
|
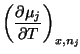 |
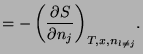 |
7.1.2 Entalpía
Es la transformada de Legendre considerando como variable activa la variable de deformación, por tanto tenemos que:
La ecuación de Euler correspondiente a este potencial es:
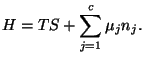
La ecuación de Gibbs-Duhem es:
Y las correspondientes relaciones de Maxwell son:
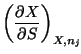 |
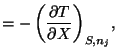 |
|
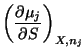 |
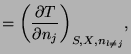 |
|
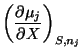 |
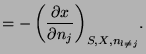 |
7.1.3 Potencial de Gibbs (entalpía libre)
Es la transformada de Legendre considerando como variables activas la entropía y la variable de deformación, por tanto tenemos que:
La ecuación de Euler correspondiente a este potencial es:
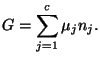
La ecuación de Gibbs-Duhem es:
Y las correspondientes relaciones de Maxwell son:
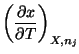 |
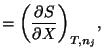 |
|
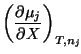 |
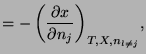 |
|
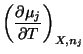 |
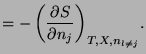 |
7.2 Ecuaciones
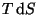
En un sistema simple y cerrado podemos expresar la entropía y la energía interna en función de dos variables, bien ![]() ,
, ![]() ó
ó ![]() . Si expresamos la entropía como
. Si expresamos la entropía como ![]() obtenemos:
obtenemos:
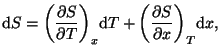
que, al aplicar una relación de Maxwell del potencial de Helmholtz nos queda en:
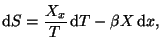
y multiplicando por
Operando de un modo similar, pero empleando una relación de Maxwell del potencial de Gibbs llegamos a la ecuación
![]() en
en ![]() :
:
Por último tenemos la ecuación
![]() en
en ![]() :
:
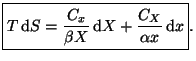
8 Tercer principio de la termodinámica
Cada vez avanza más la investigación a bajas temperaturas y es de gran interés conocer las propiedades termodinámicas de los sistemas en ese rango de temperaturas.
Experimentalmente se comprueba que cuanto más se enfría un sistema más difícil es seguirlo enfriando, de modo que el cero absoluto de temperaturas (![]() ) es prácticamente inalcanzable. Este resultado no se puede justificar a partir del primer o del segundo principio de la termodinámica, por lo que se introduce un tercero.
) es prácticamente inalcanzable. Este resultado no se puede justificar a partir del primer o del segundo principio de la termodinámica, por lo que se introduce un tercero.
La necesidad del tercer principio empezó a ser evidente cuando los investigadores intentaban obtener la afinidad química de un sistema (una variable termodinámica que nos indica cuándo dos sustancias reaccionan entre si). Al trabajar con ella se encontraban con que ésta quedaba indeterminada por una constante tras aplicar los principios primero y segundo y, por tanto, debían calcular otros parámetros del sistema.
8.1 Enunciado de Nernst
Fue Nernst quien enunció el tercer principio de la forma siguiente:
Todo sistema condensado en equilibrio estable intrínsecamente que sufra un proceso isotérmico experimenta una variación de entropía que tiende a cero a medida que la temperatura de trabajo tiende a.
Matemáticamente esto se expresa como:
Es decir, cuando nos aproximamos al cero absoluto la entropía del sistema se hace independiente de las variables que definen el estado del mismo.
Fue Planck quien le dio un valor a la constante ![]() . Esta igualdad es el enunciado de Planck del tercer principio y se propuso para que la ecuación de Boltzmann de la mecánica cuántica fuese consecuente con la expresión de la entropía.
. Esta igualdad es el enunciado de Planck del tercer principio y se propuso para que la ecuación de Boltzmann de la mecánica cuántica fuese consecuente con la expresión de la entropía.
8.2 Consecuencias físicas
He aquí algunas consecuencias sobre coeficientes termodinámicos fácilmente demostrables al expresarlos como dependientes de la entropía.
8.2.1 Capacidades caloríficas
lo cual está en contradicción con las conclusiones de la mecánica estadística clásica, que da valores constantes para los sólidos. La justificación de la expresión se da en mecánica cuántica.
8.2.2 Coeficientes térmicos
8.2.3 Entalpía y entalpía libre
A medida que
![]() obtenemos:
obtenemos:
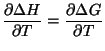 |
8.3 Cálculo de entropías absolutas
El tercer principio de la termodinámica permite obtener valores absolutos de la entropía mediante la siguiente expresión:
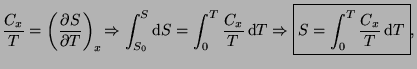
ya que
8.4 Inaccesibilidad del cero absoluto
Si queremos pasar de un punto de coordenadas termodinámicas
![]() a otro de coordenadas
a otro de coordenadas
![]() observamos que, debido a:
observamos que, debido a:
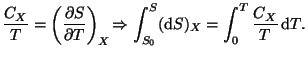
Tenemos que para los dos puntos anteriores:
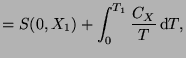 |
||
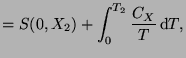 |
donde se tiene que dar que
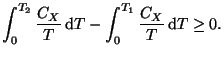
Si hacemos que ![]() , es decir, intentamos alcanzar el cero absoluto, obtenemos:
, es decir, intentamos alcanzar el cero absoluto, obtenemos:
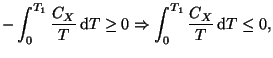
pero
A. Relaciones entre derivadas parciales
Existen dos teoremas matemáticos muy sencillos que se emplean frecuentamente en termodinámica.
Supongamos que tenemos una función que relaciona entre si las coordenadas ![]() ,
, ![]() y
y ![]() de modo que
de modo que
![]() , entonces podemos despejar una de las variables como función de las otras dos. Despejando
, entonces podemos despejar una de las variables como función de las otras dos. Despejando ![]() e
e ![]() obtenemos:
obtenemos:
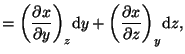 |
||
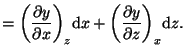 |
Por sustitución de la segunda ecuación en la primera tenemos:
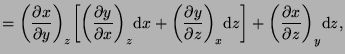 |
||
![$\displaystyle =\left(\frac{\partial x}{\partial y}\right)_{z}\!\left(\frac{\par...
...ac{\partial x}{\partial z}\right)_{y}\!\right]\mathop{\mathrm{d}\!}\nolimits z.$](img275.png) |
Por lo tanto es evidente que se tienen que cumplir las siguientes dos propiedades:
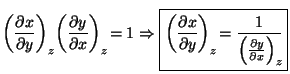
y
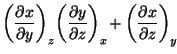 |
||
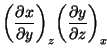 |
y por tanto obtenemos la segunda relación entre las derivadas parciales:
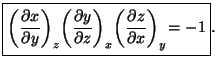
B. Transformación dual de Legendre
Sea una función
![]() dependiente de dos conjuntos de variables. Las variables
dependiente de dos conjuntos de variables. Las variables ![]() son las denominadas variables activas y las variables
son las denominadas variables activas y las variables ![]() son las denominadas variables pasivas.
son las denominadas variables pasivas.
Ahora admitimos la existencia de las derivadas parciales de la función respecto a las variables activas y denominamos a esas derivadas parciales
![]() , y suponemos también que el jacobiano es distinto de cero (es decir, que las
, y suponemos también que el jacobiano es distinto de cero (es decir, que las ![]() son independientes entre si). Esto nos permite expresar las variables dependientes como
son independientes entre si). Esto nos permite expresar las variables dependientes como
![]() .
.
Definimos ahora la transformada de Legendre como:
Ahora diferenciamos la función de forma genérica y explícita:
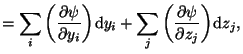 |
||
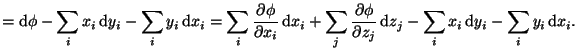 |
Igualando los ambos resultados obtenemos las siguientes igualdades:
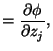 |
||
B..1 Potenciales termodinámicos
Ahora aplicamos la transformación dual de Legendre a la energía interna de un sistema en lenguaje energía
![]() donde
donde ![]() ,
,
![]() y
y
![]() .
.
Suponiendo la existencia de
![]() podemos expresar la energía infinitesimal como
podemos expresar la energía infinitesimal como
![]() , ecuación llamada ecuación de Euler. Como la energía interna es función de estado podemos aplicar el teorema de Euler y obtener la ecuación
, ecuación llamada ecuación de Euler. Como la energía interna es función de estado podemos aplicar el teorema de Euler y obtener la ecuación
![]() , llamada ecuación de Gibbs.
, llamada ecuación de Gibbs.
Definimos como potencial termodinámico ![]() a la transformada de Legendre
a la transformada de Legendre
![]()
B..2 Relaciones de Maxwell
A partir de las relaciones entre las derivadas parciales de la transformada de Legendre respecto a las variables de las que dependía podemos establecer las siguientes igualdades:
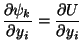 |
||
Ahora calculando las derivadas parciales segundas cruzadas e igualando obtenemos las denominadas relaciones de Maxwell:
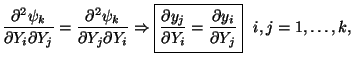
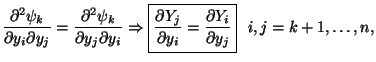
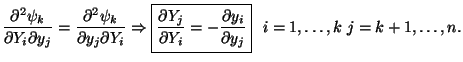

Notas al pie
- ... equilibrio1
- Decimos que un sistema termodinámico est´ en equilibrio si sus variables de estado permanecen constantes, su composición química es uniforme, es homogéneo en su composición y las variables intensivas toman el mismo valor en todo punto del sistema.
- ... calor2
- Para una definición de calor v´ase la sección 4.3
- ... caloríficos3
- Sistemas en equilibrio lo suficientemente grandes como para que al extraer o ceder una cantidad finita de calor no cambie su temperatura
- ... simples4
- Es decir, homogéneos, isótropos, químicamente inertes, sin carga, sin efectos de superficie, etc.
- ... actualidad5
- A partir de la convención de 1954
- ... escala6
- Comnmente se obtienen diferentes escalas para cada tipo de termómetro
- ... determinada7
- Sea ésta debida a un campo eléctrico, un campo magnético, un campo gravitatorio o uno cualquiera
- ... realizado8
- Como las energías potenciales gravitatoria o electrostática
- ... demostrar9
- Y en cualquier libro de termodinámica está la demostración.
- ... mismo10
- Véase la similitud con el caso de la energía interna
